How to use R to rewrite FCS files with different number of channels
Background
Often during file acquisition at the cytometer, you may acquire FCS files with different numbers of channels even though they have the same staining panels. When the number of channels is mismatched between files that otherwise use the same panel, it will impose challenges for downstream analysis, especially for the advanced machine learning algorithmic analysis. This technical note can be used as step-by-step guidance on how to fix the panel and channel mismatch issue by using the R cytofCore package.
Objectives
- Learn how to rewrite FCS files that have different number of channels.
Prerequisite steps for running the R scripts
- Install the latest version of R to your computer http://www.r-project.org/
- Install RStudio http://www.rstudio.com/
- Install flowCore library http://www.bioconductor.org/packages/release/bioc/html/flowCore.html
- Install cytofCore library https://github.com/nolanlab/cytofCore#Installation
- For Macintosh operating system, install this software, which requires logging out and logging back in on your computer account to start working. http://xquartz.macosforge.org/landing/
How to rewrite FCS files that have different number of channels.
If you have FCS files that have different number of channels and need to unify the channel number, you can run the following script. Channels can either be added or removed based on a template file that has the desired number and order of channels. The template file can be an existing FCS file.
All the files that need channels updated must be in one folder and the template file must be in a separate path. After running the last line below, you will be prompted to choose the template file and the file directory (Figure 1a and 1b). Select the template file and click on open, then select the directory of your target FCS files that need to be updated. When choosing the folder, you must open that folder in the selection window before clicking OK. A subfolder called ‘relabeled’ will be created within the selected folder, and new FCS files will be written there.
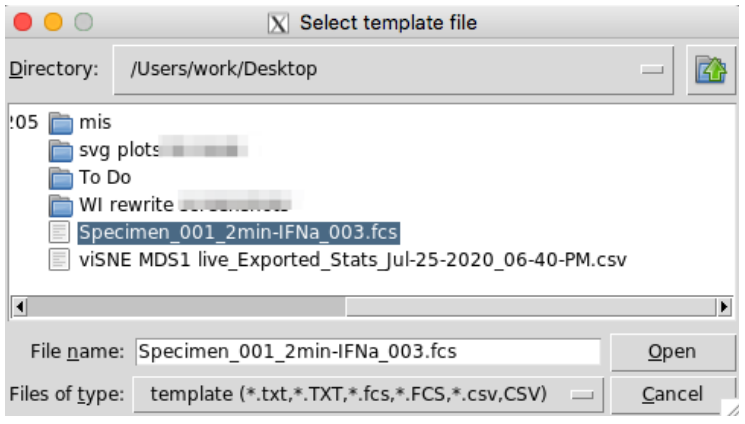
Figure 1a.

Figure 1b.
Open RStudio that was installed on your computer during the prerequisite steps and run the following commands. Please have the template file and your target files ready as described above.
install.packages(“devtools”)
install.packages(“XML”)
library(“devtools”)
if (!requireNamespace(“BiocManager”, quietly = TRUE))
install.packages(“BiocManager”)
BiocManager::install(version = “3.12”)
BiocManager::install(“flowCore”)
install_github(“nolanlab/cytofCore”)
library(“cytofCore”)
#When choosing the folder, you must enter that folder in the selection window before clicking OK
# All the files that need to update channels must be in one folder and the template file in a separate path
Tips for success
- This script will remove the compensation matrix from FCS files. If you are working with flow cytometry data, please make sure to reapply the compensation to the data before further analysis. Here is an instruction on how to import compensation in Cytobank. Or if you have access to Kaluza Analysis software, it might be easier to edit channels in Kaluza and export new FCS files using the Kaluza Cytobank Plugin. For more information visit http://www.kaluzasoftware.com/
- Please write to [email protected] for support with any issues.
- This tech note uses R 4.0.3 and Bioconductor 3.12
References
http://www.bioconductor.org/packages/release/bioc/html/flowCore.html
